Sfoglia categorie
Esplora
Fiverr Pro
Italiano
$
USD
Appassionato di apprendimento
Informazioni su questo servizio
Se lavori su un sistema proteina-ligando e hai bisogno di simulazioni di dinamica molecolare affidabili e ben analizzate per la tua ricerca o pubblicazione, offro servizi end-to-end di simulazione MD con AMBER, dalla preparazione del sistema alla creazione di figure pronte per la pubblicazione, utilizzando lo stesso pipeline impiegato in ricerche peer-reviewed di scoperta di farmaci computazionale.
Sono un ricercatore in biologia computazionale con esperienza pratica in AMBER/AmberTools, cpptraj e workflow di analisi basati su Python. Il mio lavoro ha supportato direttamente manoscritti inviati a riviste peer-reviewed in farmacologia computazionale.
Cosa consegno
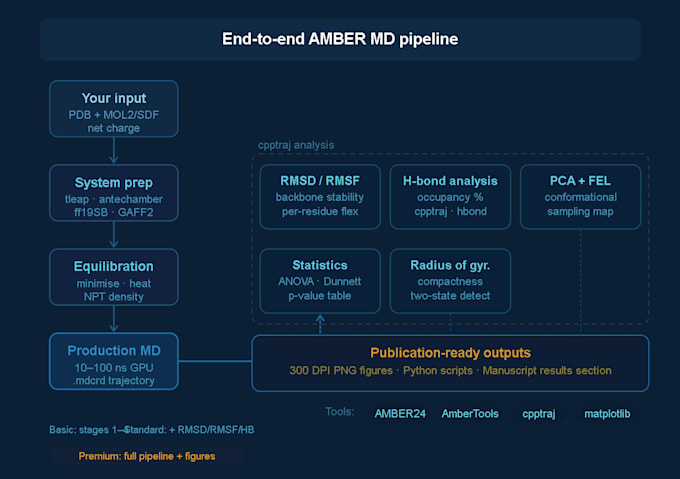
Configurazione del sistema
Preparazione della struttura proteica e assegnazione dello stato di protonazione
Parametrizzazione del ligando usando antechamber
Solvatazione (casella d'acqua TIP3P o OPC), aggiunta di controioni
Assegnazione del force field: ff19SB (proteina), GAFF2 (piccola molecola)
Minimizzazione, riscaldamento e equilibration NPT/NVT
Esecuzione della simulazione MD di produzione
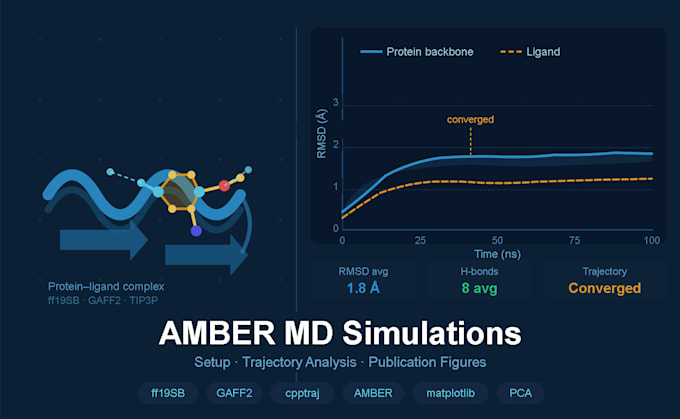
Traiettorie di produzione di 10, 50 o 100 ns (dipende dal pacchetto)
File di traiettoria .mdcrd completi consegnati
Analisi della traiettoria (Standard & Premium)
Figure pronte per pubblicazioni complete
Perché lavorare con me?
Esperienza reale con amber, non un wrapper cloud o uno strumento online.